Chest Medicine
Obstructive Lung Disease
Restrictive Lung Disease
Pulmonary Vascular Diseases
Neoplasms of the Lungs
Pleural Disease
Other Respiratory Disorders
Idiopathic pulmonary fibrosis (IPF)
- Inflammatory infiltrates of cells into the pulmonary interstitium associated with fibrosis of unknown cause
Signs and Symptoms
- Symptoms: dry cough, extertional dyspnoea, weight loss, malaise
- Signs: cyanosis, finger clubbing, fine end-inspiratory crackles
- Complications: respiratory failure, may be associated with an increased risk of lung cancer
Investigation
- Blood tests: ABG, CRP, immunoglobulins, rheumatoid factor, ANA
- CXR: reduced lung volume, bilateral reticulo-nodular shadows, may see honeycombing in end-stage interstitial disease
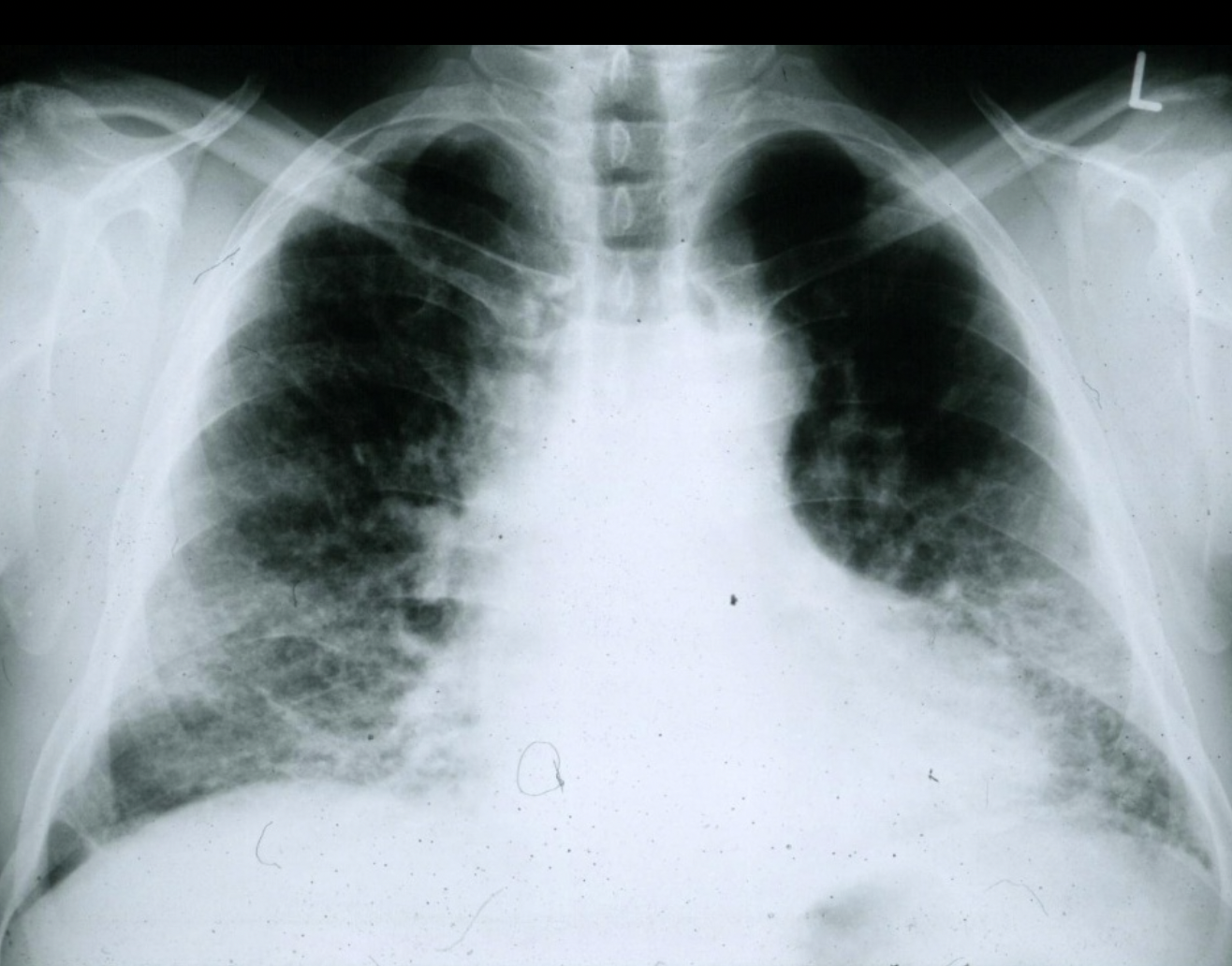
A chest radiograph of a patient with IPF. Note the small lung fields and peripheral pattern of reticulonodular opacification. (courtesy of wikipedia.org) - High-resolution CT chest:
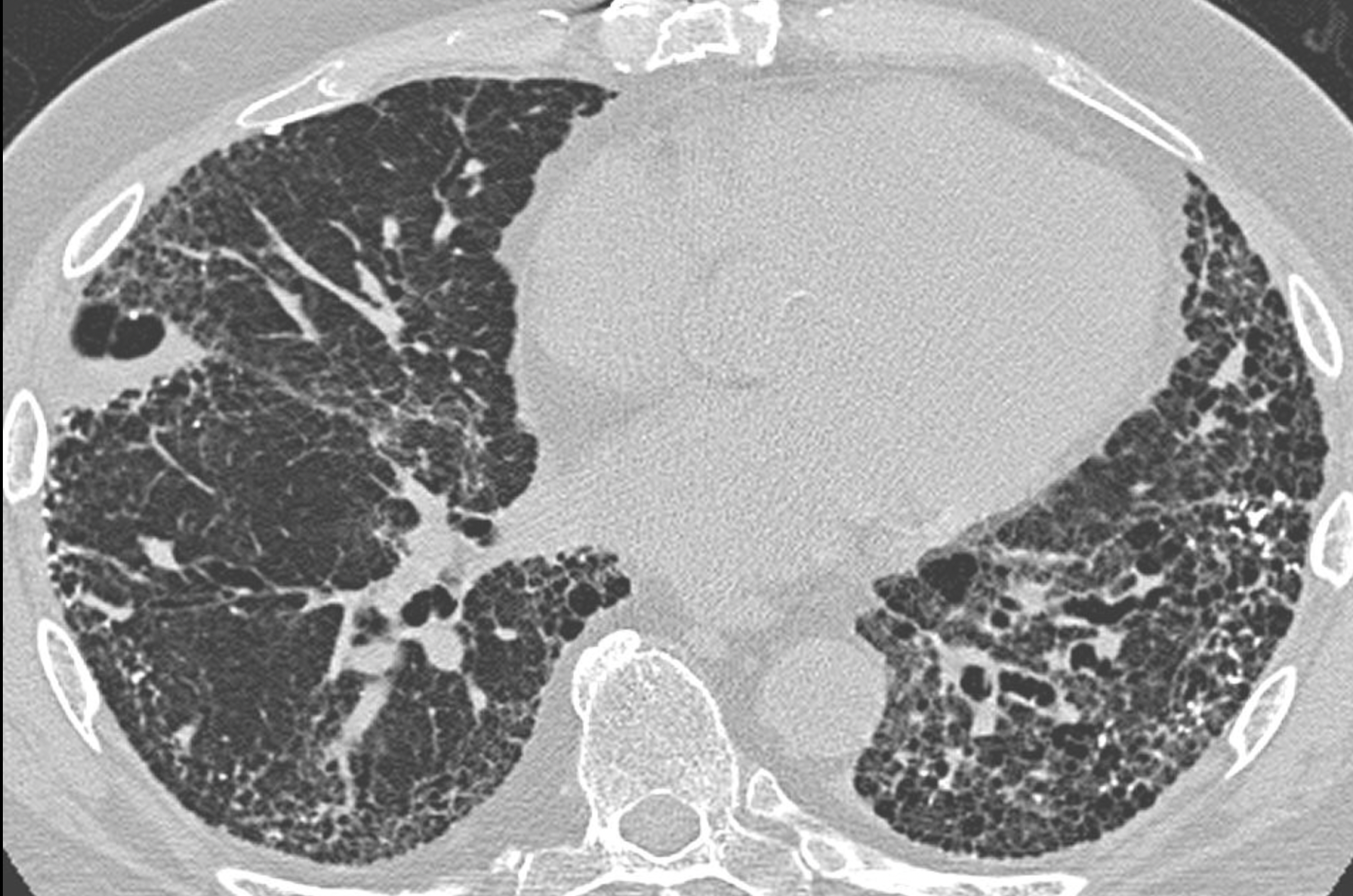
- UIP (usual interstitial pneumonia) pattern: honeycombing (> 5% of lung parenchyma), reticular opacity (dominant) and ground-glass opacity (less extensive than reticular pattern) → carries a poor prognosis (50% survival at 3-4 years)
- NSIP (Non-specific interstitial pneumonia) pattern: bilateral ground glass opacity (dominant), reticular opacity (less extensive), usually absence of honeycombing → carries a better prognosis (divided into fibrotic and cellular subtypes → 90% survival at 5 years if cellular type or 60% at 5 years if fibrotic type)
- Spirometry: restrictive pattern
- BAL: may show alveolitis (lymphocytosis, neutrophilia and/or eosinophilia)
- Lung biopsy: gives histological diagnosis
Management
- Supportive care: O2, pulmonary rehabilitation, palliative care
- Anti-fibrotic agents: nintedanib, pirfenidone
- Lung transplant
- High-dose steroids are not used unless diagnosis of IPF is doubtful
Reference: Oxford Handbook of Clinical Medicine (10th Edition)